
近日,我所化学反应动力学全国重点实验室团簇光谱与动力学研究组(2506组)江凌研究员、谢华研究员团队利用自主研制的质谱-光电子能谱实验方法,研究了金属碳化物团簇MC3-(M = Os,Ir,Pt)的固氮模式,揭示了直接裂解N≡N三键与化学吸附双重固氮模式的竞争机制,为开发高效固氮催化剂和单原子材料提供了新思路。
氮气分子由于其极强的N≡N三键(键能约为9.79 eV),在温和条件下难以被活化。当前,工业固氮依赖Haber-Bosch法,需在高温高压下进行,能耗巨大。金属碳化物作为一类高潜力的催化材料,因其反应机制尚不明确,其实际应用长期受限。本项研究通过质谱-光电子能谱实验方法与量子化学计算方法相结合,阐明了金属碳化物团簇与氮气的反应路径,揭示了金属碳化物活化氮气的两种重要竞争机制。
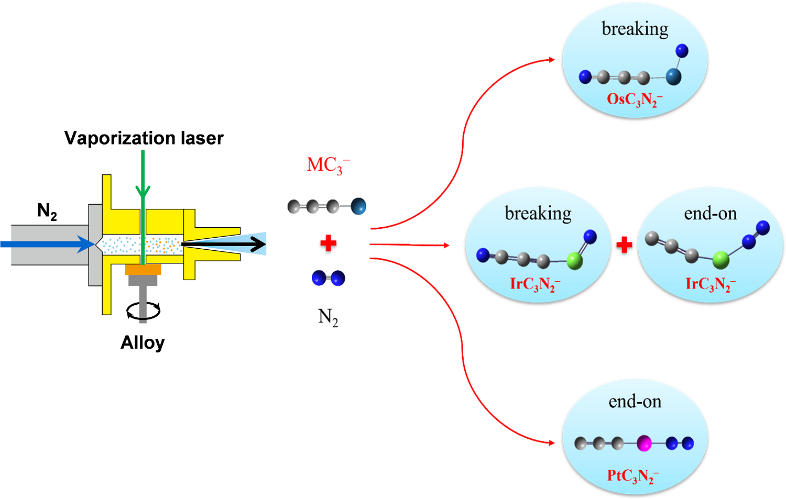
江凌和谢华研究团队长期致力于金属团簇与资源小分子(例如N2、CO等)的成键和反应机制的研究。在前期相关研究(J. Phys. Chem. Lett.,2024;Angew. Chem. Int. Ed.,2023;JACS Au,2023;Inorg. Chem.,2023;J. Phys. Chem. Lett.,2022;J. Energy Chem.,2021)的基础上,本工作中团队采用激光溅射团簇源的方式产生了MC3N2− (M= Os,Ir,Pt)负离子,并获得其丰富的电子结构信息。研究发现,IrC3⁻可同时通过裂解和吸附的两种机制活化氮气,OsC3⁻更偏好于直接断裂N≡N键生成C-N键, 而PtC3⁻则倾向化学吸附。理论计算进一步表明,金属5d轨道能量是决定反应活化路径的关键因素:5d轨道能量越高,越易触发N≡N键断裂;反之则化学吸附占主导。这一规律与实验结果高度吻合,为定向设计金属中心活性位点提供了量化依据。该研究不仅在原子尺度揭示了金属碳化物的反应特性,也为开发具有孤立贵金属中心的单原子催化剂提供了新的策略。
相关研究成果以“Observation of competing nitrogen activation in metal tricarbon anions MC3– (M= Os,Ir,Pt)”为题,发表在《化学科学》(Chemical Science)上。该工作的共同第一作者是我所2506组已毕业的联合培养硕士杜世虎和张子恒。该工作得到科技部科技创新2030-重大项目、国家自然科学基金、中国科学院青促会、我所创新基金等项目的资助。(文/图 杜世虎)


